
II. Background
- Thalassemia is derived from Greek word "thalassa" for sea
III. Epidemiology
- Thalassemia accounts for one third of all globin abnormalities
- Gender: Males and females affected equally
-
Prevalence of Thalassemia
- Among at risk ethnicities: 5-30%
- North and South America: 6 per 100,000 conceptions
- World wide
- Alpha Thalassemia: 5% worldwide Prevalence
- Beta Thalassemia: 1.5% worldwide Prevalence
- Ethnicity
- Alpha Thalassemia
- Beta Thalassemia
- Southern Italy and Mediterranean islands (0.1% Incidence)
- Central Africa
- Southeast Asia
IV. Pathophysiology
- Thalassemia is a cluster of Autosomal Recessive hematologic disorders affecting Hemoglobin
- Globin chain (alpha or beta) abnormalities resulting in Anemia with decreased Hemoglobin A
- Unbalanced red cells that are susceptible to Hemolysis
- Ineffective Erythropoiesis
- Images
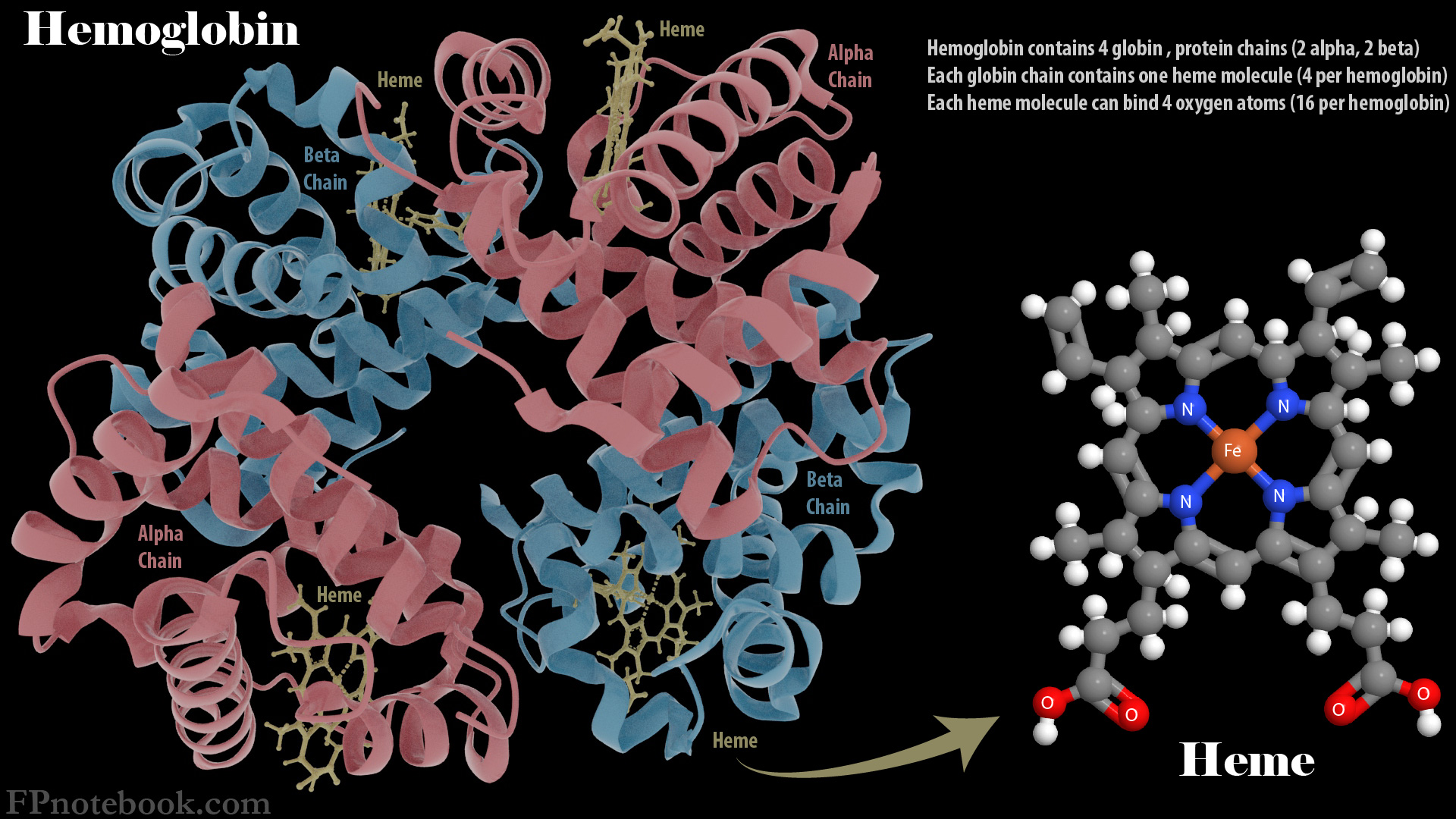
V. Types: Based on Hemoglobin Defect
-
Alpha Thalassemia
- Asymptomatic
- Alpha Thalassemia Silent Carrier (Alpha Thalassemia Minima)
- Alpha Thalassemia Trait (Alpha Thalassemia Minor)
- Moderate to Severe (HbH Disease)
- Alpha Thalassemia Intermedia (Deletional HbH Disease)
- Hemoglobin Constant Spring (Non-Deletional HbH Disease)
- Very Severe
- Asymptomatic
-
Beta Thalassemia
- May also be combined with other Hemoglobinopathy (HbC, HbE, HbS)
- Asymptomatic
- Moderate to Severe
VI. Types: Based on Transfusion Dependence
- Transfusion-Dependent Thalassemia (TDT)
- Beta Thalassemia Major (Cooley's Anemia)
- Hemoglobin Constant Spring (Non-Deletional HbH Disease)
- Survived Alpha Thalassemia Major (Hemoglobin Bart, Non-Immune Hydrops Fetalis)
- Non-Transfusion Dependent Thalassemia (NTDT)
- Asymptomatic Thalassemia (silent carrier or trait)
- Intermittent transfusions may be required
VII. Symptoms: Presentations
- Typically asymptomatic for carrier and trait states
- Moderate to severe Microcytic Anemia
- Fatigue
- Dyspnea
- Light Headedness or Near Syncope
- Growth Delay in children
- Hemolytic Anemia
- Chronic Anemia in Children may result in Erythropoietin induced changes
- Extramedullary hematopoiesis
- Bone Marrow expansion results in bony deformities of facial and extremity long bones
- Frontal Bossing
- Maxillary Hypertrophy
- Malar prominence
- Bone masses
VIII. Labs: Red Cell Indices and Iron Sudies
-
Complete Blood Count
- Hemoglobin or Hematocrit consistent with Anemia
-
Mean Corpuscular Volume (MCV)
- Hematocrit >30% and MCV low but >80 fl: Iron Deficiency Anemia more likely
- Hematocrit >30% and MCV <75 fl: Thalassemia more likely
- However MCV cut-off suggestive of Thalassemia varies by age
- MCV <70 fl up to 6 years
- MCV <75 fl in age 7-12
- MCV <80 fl in adults
-
Red Cell Distribution Width (RDW)
- Microcytosis with Normal RDW
- Thalassemia is most likely
- Microcytosis with Increased RDW
- Sideroblastic Anemia
- Iron Deficiency Anemia (typically RDW >15%)
- Thalassemia (RDW can be high, esp. in Beta Thalassemia)
- Microcytosis with Normal RDW
-
Mean Corpuscular Volume to Red Blood Cell Count ratio (applies to evaluation in children)
- See Mentzer Index
- Ratio <13: Thalassemia
- Ratio >13: Iron Deficiency Anemia, Hemoglobinopathy
- Normal Iron study indices (no Iron Deficiency Anemia)
- Serum Ferritin normal or elevated (often >100 ng/ml)
- Serum Ferritin >12 ng/ml favors Thalassemia (outside of inflammatory states)
- Serum Ferritin <12 ng/ml favors Iron Deficiency Anemia
- Other iron studies typically not needed unless inflammation is present
- Total Iron Binding Capacity normal
- Serum Iron normal
- Serum Ferritin normal or elevated (often >100 ng/ml)
- Variable Reticulocyte Index
- May see Reticulocytosis or Reticulocytopenia
IX. Labs: Peripheral Smear
- Hypochromic, microcytic red cells
- Poikilocytosis (irregularly shaped red cells)
- Typical for Thalassemia (esp. Beta Thalassemia)
- May be seen in severe Iron Deficiency Anemia
-
Hemolytic Anemia signs (Target Cells, Red Blood Cell Inclusion bodies)
- Typical for Thalassemia (esp. Beta Thalassemia)
X. Labs: Hemoglobin Electrophoresis (Hgb Electrophoresis)
- Hemoglobin Electrophoresis is required for Thalassemia diagnosis
-
Iron Deficiency
- Normal Hemoglobin Electrophoresis
- HbA2 may be low
-
Alpha Thalassemia
- Adult carrier or Alpha Thalassemia Trait
- HbA Normal at >95%
- HbA2 Normal at 2 to 3.5%
- HbF Normal at <1%
- HbH Absent (normal)
-
Alpha Thalassemia Intermedia (HbH Disease)
- HbA below normal
- HbA2 <4%, but typically below normal
- HbF Abnormal at 5 to 25%
- HbH Abnormal at 0.8 to 40% (key finding)
- Newborns
- HbH or Hb Bart may be present
- Adult carrier or Alpha Thalassemia Trait
-
Beta Thalassemia
-
Beta Thalassemia Trait
- HbA Normal at >90%
- HbA2 Increased at 3.5 to 9%
- HbF may be increased to up to 5%
- HbH Absent (normal)
-
Beta Thalassemia Major
- HbA decreased or absent
- HbA2 increased at >4%
- HbF increased to 10 to 50% (may be as high as 100%)
-
Beta Thalassemia Trait
XI. Labs: Other
-
Genetic Testing
- Confirms Thalassemia
- Identifies specific mutations (and predicts associated severity)
-
Genotype and HLA Typing
- Obtained in all patients (esp. age <12 years old, see management below)
- Used in those being considered for Hematopoietic Stem Cell Transplant or gene therapy
XII. Differential Diagnosis
- See Microcytic Anemia
- See Hemolytic Anemia (esp. Beta Thalassemia)
XIII. Imaging
- MRI
- Identifies degree of Iron Overload
- Obtain T2 weighted Cardiac MRI or R2 weighted Liver MRI
XIV. Evaluation: Thalassemia Screening Indications
- Pregnancy and Preconception Counseling
- Obtain Complete Blood Count in all pregnant patients (and Hgb Electrophoresis if MCV low)
- Also consider Hgb Electrophoresis in high risk ethnicity (see above), Thalassemia in first degree relatives
-
Prenatal Diagnosis (in fetus of parents with Thalassemia)
- Chorionic Villus Sampling (10 to 12 weeks gestation)
- Amniocentesis (>15 weeks gestation)
-
Newborn Screening
- Thalassemia is not on the U.S. core universal Newborn Screening panel, but many states screen for Thalassemia
XV. Management: General
- See Alpha Thalassemia
- See Beta Thalassemia
- Thalassemia International Federation uses transfusion dependence more than subtypes to direct management
- Transfusion-Dependent Thalassemia (TDT)
- Non-Transfusion Dependent Thalassemia (NTDT)
XVI. Management: Blood Transfusion
- Transfusion Dependent Indications
- Hemoglobin <7 g/dl (70 g/L) OR
- Anemia Complications (at least one)
- Growth Delay or Delayed Puberty
- Anemia related functional limitations (Fatigue impacting school or work, low Exercise tolerance or quality of life)
- Erythropoietin-Induced Changes
- Extramedullary hematopoiesis (Hepatosplenomegaly)
- Bone Marrow expansion (e.g. Frontal Bossing, Maxillary Hypertrophy, Malar prominence)
- Target Hemoglobin in Transfusion Dependent Thalassemia
- Pretransfusion Hemoglobin 9 to 10.5 g/dl (90 to 105 g/L)
- Posttransfusion Hemoglobin up to 11 to 12 g/dl (110 to 120 g/L)
- Protocol in Transfusion Dependent Thalassemia
- Transfusions may be needed as early as 6 months of age
- Transfusion scheduled every 2 to 5 weeks
- Risk of Transfusion Reaction, alloimmunization, bloodborne infection, Iron Overload
- Iron chelation often used in combination in those over age 2 years, after first 10-12 transfusions (or otherwise indicated)
- Folic Acid supplementation is used in Transfusion Dependent Thalassemia
- Intermittent transfusion indications in Non-Transfusion Dependent Thalassemia
- Symptomatic Anemia
- Pregnancy
- Preoperative state
- Serious infections
XVII. Management: Iron Chelation
- Thalassemia increases the risk of Iron Overload (frequent transfusion, Hemolytic Anemia, increased GI iron absorption)
- Iron Overload complications (liver, heart) are reduced with early initiation of chelation therapy
- Indications (age >2 years old, at least one criteria)
- Number of transfusions >10 to 12
- High Serum Ferritin
- Transfusion-Dependent Thalassemia (TDT): Serum Ferritin > 1000 ng/ml (or mcg/L)
- Non-Transfusion Dependent Thalassemia (NTDT): Serum Ferritin >800 ng/ml (or mcg/L)
- MRI demonstrating Iron Overload
-
Iron chelators
- Deferoxamine (Desferal) SQ/IV
- Deferasirox (Exjade) Orally
XVIII. Management: Other Measures
-
Hydroxyurea
- Stimulates Hemoglobin F synthesis
- May reduce transfusion frequency in Beta Thalassemia Intermedia and Beta Thalassemia Major
- Luspatercept (Reblozyl)
- Activin Receptor Ligand Trap
- Increases Erythropoiesis and decreases transfusion frequency in Beta Thalassemia
-
Hematopoietic Stem Cell Transplant (Bone Marrow Transplantation)
- Curative of transfusion dependent Beta Thalassemia when performed in childhood in low risk patients
- Preferred patient is <12 years old with HLA matched sibling donor
-
Gene Therapy (experimental as of 2022)
- Consider in patients over age 12 years
- Targets increasing normal beta globin synthesis or reactivating Hemoglobin F synthesis
XIX. Complications: Anemia Related
- Infants and children
- Growth Delay
- Delayed Puberty
- Erythropoietin-Induced Bone Marrow expansion (e.g. Frontal Bossing, Maxillary Hypertrophy, Malar prominence)
- Anemia related functional limitations
- Hypersplenism
- Causes
- Splenic hyperfunction to remove defective Red Blood Cells
- Erythropoietin-Induced extramedullary hematopoiesis
- Splenectomy
- Improves baseline Hemoglobin (by 1 to 2 g/dl) and reduces transfusion frequency
- Risk of Asplenia (infection, Venous Thromboembolism, Pulmonary Hypertension)
- Indications (age >5 years)
- Iron Overload refractory to iron chelation
- Hypersplenism with Clinically Significant cytopenias
- Symptomatic Splenomegaly
- Causes
- Venous Thrombosis (especially after splenectomy)
- Most common with Beta Thalassemia Major and Intermedia
- Consider Perioperative Anticoagulation
- Thromboprophylaxis is recommended in pregnancy
- Avoid exacerbating Hypercoagulable state (e.g. avoid oral contarceptives in women)
-
Osteoporosis and Osteopenia
- Seen in up to 50% of Beta Thalassemia Major, even with transfusions and iron chelation
- Encourage Osteoporosis Prevention (e.g. Physical Activity, Calcium Supplementation, Vitamin D Supplementation
- Consider hormonal therapy, Bisphosphonates and zinc supplementation
XX. Complications: Iron Overload Related (Transfusion Dependent Thalassemia)
- Endocrine Disorders (decreased with chelation therapy)
- Diabetes Mellitus (Iron Overload related)
- Hypogonadotropic Hypogonadism
- Hypothyroidism
- Hypoparathyroidism
- Growth Hormone Deficiency (8 to 14% of Transfusion Dependent Thalassemia)
- Amenorrhea (primary or secondary)
- Cardiac Disorders (decreased with iron chelation)
- Associated with cardiac Iron Overload (seen in 25% of Beta Thalassemia worldwide)
- Myocardial fibrosis
- Cardiomyopathy
- Heart Failure
- Pulmonary Hypertension
- Cardiac Dysrhythmia
- Valvular heart disease
- Pericarditis
- Myocarditis
-
Liver Disease (decreased with iron chelation)
- Cirrhosis (2 to 7% of Transfusion Dependent Thalassemia)
- Hepatocellular Carcinoma (<3.5%)
- Hepatitis C risk increases due to frequent transfusion
XXI. Prognosis
- Normal Lifespan
- Asymptomatic Thalassemia (trait, carrier)
- Alpha Thalassemia Intermedia (Deletional HbH Disease)
- Possibly Reduced Lifespan
- Hemoglobin Constant Spring (Non-Deletional HbH Disease)
- Associated with increased transfusions and related complications
- Hemoglobin Constant Spring (Non-Deletional HbH Disease)
- Reduced Lifespan
- Beta Thalassemia Major
- Average lifespan to age 50 years
- Improved from prior 17-30 year lifespan with transfusions and prevention of Iron Overload
- Beta Thalassemia Minor
- Average lifespan to age 57 years
- Average lifespan also improved from prior with transfusions and prevention of Iron Overload
- Beta Thalassemia Major
- Neonatal mortality
- Alpha Thalassemia Major (Hemoglobin Bart, Non-Immune Hydrops Fetalis)
- Had been uiniformy fatal before the use of intrauterine transfusions
- Intrauterine transfusions have allowed for initial survival approacing 100%
- Intrauterine Bone Marrow Transplantation is being explored as of 2022
- Alpha Thalassemia Major (Hemoglobin Bart, Non-Immune Hydrops Fetalis)
XXII. Prevention
- See Thalassemia
- Preconception Genetic Counseling for parents with Thalassemia
- Chorionic Villus Sampling can diagnose Thalassemia in first trimester
- Preimplantation Genetic Testing can predict Thalassemia prior to in vitro fertilization